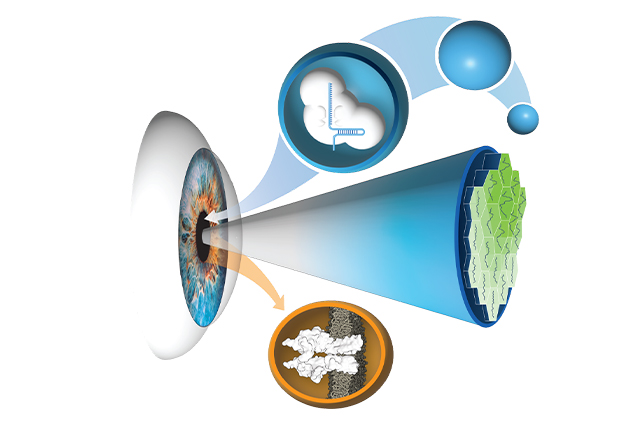
Pattnaik, who holds a doctorate in biophysics and is affiliated with the Department of Ophthalmology and Visual Sciences, led a team including scientists and engineers from UW–Madison, Harvard, and MIT that published the study in the Journal of Clinical Investigation. The researchers are seeking to develop treatments for two forms of childhood blindness caused by retinal gene mutations, LCA and Best disease, which is also known as Best vitelliform macular dystrophy. LCA begins in infancy and causes extreme farsightedness, sensitivity to light, and involuntary eye movements. Best disease, usually diagnosed in childhood, causes macular degeneration and loss of central vision, visual acuity, and color perception.
The diseases are relatively rare, affecting 3 to 4 individuals per 100,000 people, but the research team expects that its new nanoparticle-packaged CRISPR technology will be able to treat other inherited eye diseases.
An innovative technique distinguishes the work from a more typical method used in gene editing. Since the CRISPR gene editing method known as CRISPR/Cas 9 was discovered in 2012, using a modified virus has become the norm for delivering gene-correcting material. However, the virus transport method has disadvantages in its degree of precision, the number of cells affected, and the possibility of unpredictable side effects.
The team reasoned that silica nanocapsules do not interact with the body, lowering the risk of problematic and unpredictable immune system responses that viral methods may elicit. Another advantage of the technique is its specificity. “The rapid pace of genetic diagnosis development has enabled accuracy in identifying disease-causing mutations, and we used base editing to correct a specific error in the DNA sequence of patients, overcoming several challenges through the collaborative team science approach,” said research associate Meha Kabra.