Health & Wellness
July 8, 2026
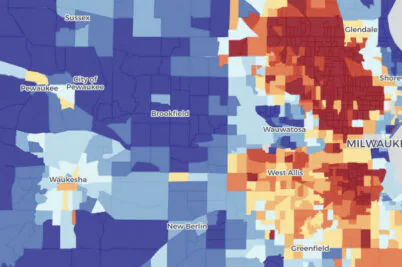
Milwaukee taps UW–Madison research to inform lead pipe replacement strategy
In Milwaukee, tens of thousands of households still receive drinking water through lead service lines. The pipes were installed many decades before medical research made clear that even a small amount of lead is toxic, particularly for small children.